

What Informed Consent Really Asks of Patients
Why understanding a clinical trial isn’t a single moment — and can’t be reduced to a form

The room is quiet in a way that feels heavier than silence.
It smells faintly of disinfectant — that clean, sharp scent that never quite fades in medical buildings. The paper on the exam table crinkles when someone shifts their weight. A clock ticks softly somewhere out of sight.
Nothing is wrong, exactly. The chair is comfortable. The explanation has been careful. The clinician has done everything right. And still, something has slowed.

A folder is set on the table. It makes a soft sound when it lands — thicker than expected, heavier than it looks. Someone says, “You don’t have to decide today.” Someone else adds, “Take it home if you’d like.”
This is the moment people recognize as informed consent.
For many, it arrives already carrying meaning. Clinical trials are not neutral words. They’re freighted with stories — of last chances, of risk, of being experimented on rather than treated. Even when no one says these things out loud, they hover at the edges of the room.
What makes this moment different from most medical decisions isn’t fear.
It’s uncertainty.
When people consent to a common surgery or a standard treatment, the conversation is about known ground. The procedure has been done before. Recovery looks roughly the same for most people. Complications are discussed, but they are exceptions to a well-understood path.
Here, the path is still being drawn.
The information being shared is accurate, careful, and incomplete — not because anything is being hidden, but because the answers do not yet exist. The trial is happening precisely because medicine does not yet know what will work, for whom, and under what conditions.
The document in the folder tries to hold all of that at once.
It lists risks. It explains procedures. It describes alternatives. It reassures that participation is voluntary. It does what it is designed to do.
And still, the real question sits elsewhere.
Not “Do I understand what’s written here?”
But “Am I willing to step into something that is still becoming known?”
That is the question informed consent actually asks. And it doesn’t get answered all at once.
The Moment Where Everything Slows Down
In most medical encounters, time moves forward with purpose.
You describe symptoms. Someone interprets them. A plan is proposed. Even when choices are difficult, there is usually a sense of direction — a recommendation shaped by experience, precedent, and probability. Someone is carrying the weight of what comes next.
Informed consent interrupts that rhythm.
The room doesn’t change, but the language does. What will happen becomes what may happen. What is typical gives way to what is possible. The clinician’s tone stays steady, but the conversation grows careful, precise.
This is the moment when responsibility shifts.
Not entirely — clinicians and researchers remain accountable — but enough that patients feel it. The decision is no longer being carried for them. It’s being handed to them, gently, with time to consider it.
That pause is subtle. And it is heavy.
Before Anyone Asks You to Decide
Long before informed consent enters the conversation, the trial has already taken shape.
Design decisions were made months or years earlier. Someone decided which outcomes mattered most. Someone chose how often participants would need to come in, how long visits would last, which tests were essential and which were tolerable. Assumptions were made about what people could realistically manage alongside work, caregiving, fatigue, and ordinary life.
None of this is arbitrary. All of it is constrained — by science, regulation, funding, and what has worked before.
But none of it is visible to the person sitting in the room.
Consent does not ask patients to co-design the study. It asks them to enter something already built — a structure imagined for many, now offered to one.
The uncertainty isn’t whether the trial exists. It’s whether it will fit into a real life.
Why the Paper Exists — and Why It’s Never Enough
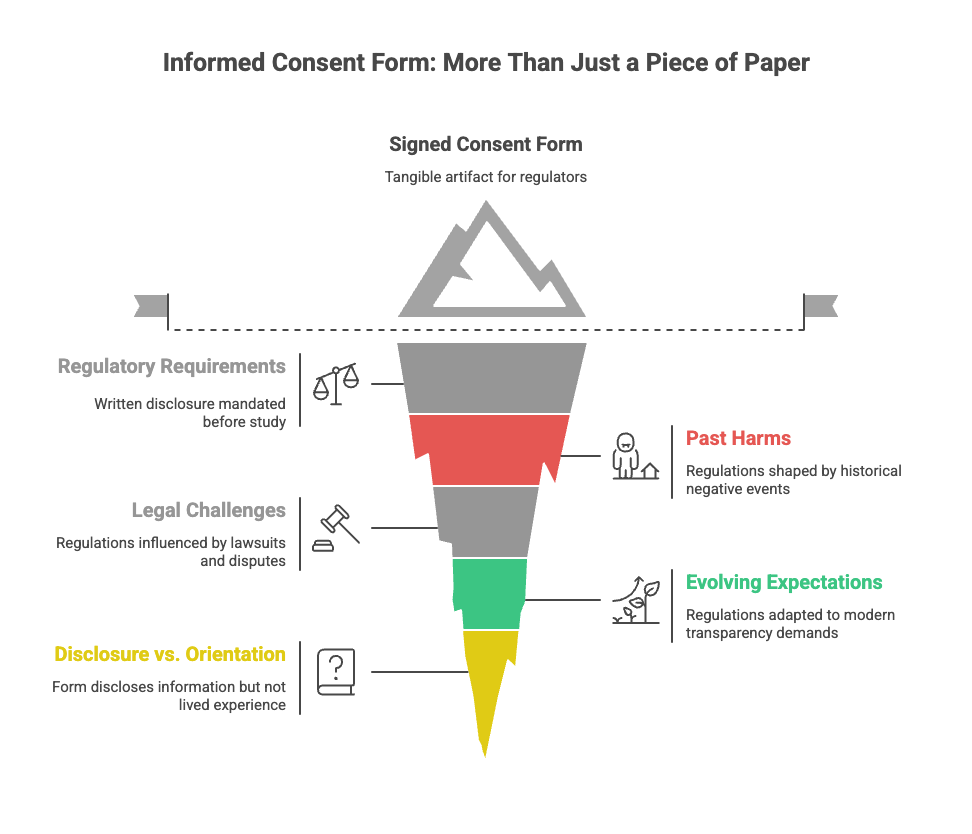
The informed consent form is often mistaken for consent itself.
That confusion is understandable. The form is tangible. It can be signed, filed, audited. It is the artifact regulators review and institutions rely on when asked whether consent occurred.
The form exists because regulations require that specific information be disclosed in writing before a clinical study can begin. Over time, those requirements have expanded — shaped by past harms, legal challenges, and evolving expectations around transparency and autonomy.
Each addition makes sense on its own.
Together, they create a document that tries to anticipate every question in advance.
The result is often a long, dense form written to ensure completeness rather than ease. It succeeds at what it is designed to do: disclose.
But disclosure is not the same thing as orientation.
No document can tell someone how participation will feel once it becomes part of their routine. No page can prepare them for how uncertainty stretches when timelines shift or expectations change.
The paper matters. It just isn’t where understanding finishes.
How Clinical Trials Differ From Standard Medical Care
Most people intuitively understand how consent works for routine medical care.
If you are scheduled for a common surgery — a knee replacement, a gallbladder removal — the consent conversation is about known risks and expected outcomes. The procedure has been done many times before. Recovery timelines are familiar. The range of what usually happens is relatively narrow.
A knee replacement does not change halfway through the operation.
Clinical trials are different.
They exist precisely because that level of certainty does not yet exist. The intervention — whether a drug, a device, or a procedure — is still being studied. The dose may not be optimized. Long-term effects may not be known. How different bodies respond is still being learned.
In a trial, change is not a deviation. It is the point.
Doses are adjusted because learning is happening. Devices are modified because performance is still being understood. Procedures evolve because early assumptions are being tested in real bodies, not simulations.
This distinction matters when we talk about consent.
In standard care, consent is about permission.
In research, consent is about participation in learning.
How Understanding Changes as Knowledge Changes
Informed consent often assumes that information is stable. In clinical research, it isn’t.
Trials exist because answers are incomplete. The information shared at enrollment is accurate — but provisional. As the study unfolds, knowledge accumulates. Patterns emerge. Some questions narrow. Others widen.
Understanding doesn’t simply deepen. It shifts.
What felt abstract at enrollment becomes concrete months later. What seemed unlikely begins to feel possible. What wasn’t yet known becomes relevant — sometimes urgently.
Consent is revisited not because people failed to understand the first time, but because the landscape itself has changed.
The temperature changes because the system is learning in real time.
What Participation Feels Like From the Inside
When patients agree to participate in a trial, they are not just agreeing to procedures.
They are agreeing to reorganize parts of their lives.
Calendars tighten. Routines shift. Ordinary days now carry appointments, travel, recovery time. Even in well-run trials, participation introduces friction — small, cumulative disruptions that don’t show up neatly on consent forms.
This is true whether the intervention is a daily pill or a one-time device implantation. The mechanics differ. The lived experience is similar: adapting to a structure you didn’t design, while trying to live the rest of your life inside it.
Consent marks the moment someone agrees to carry that weight — without knowing exactly how heavy it will feel.
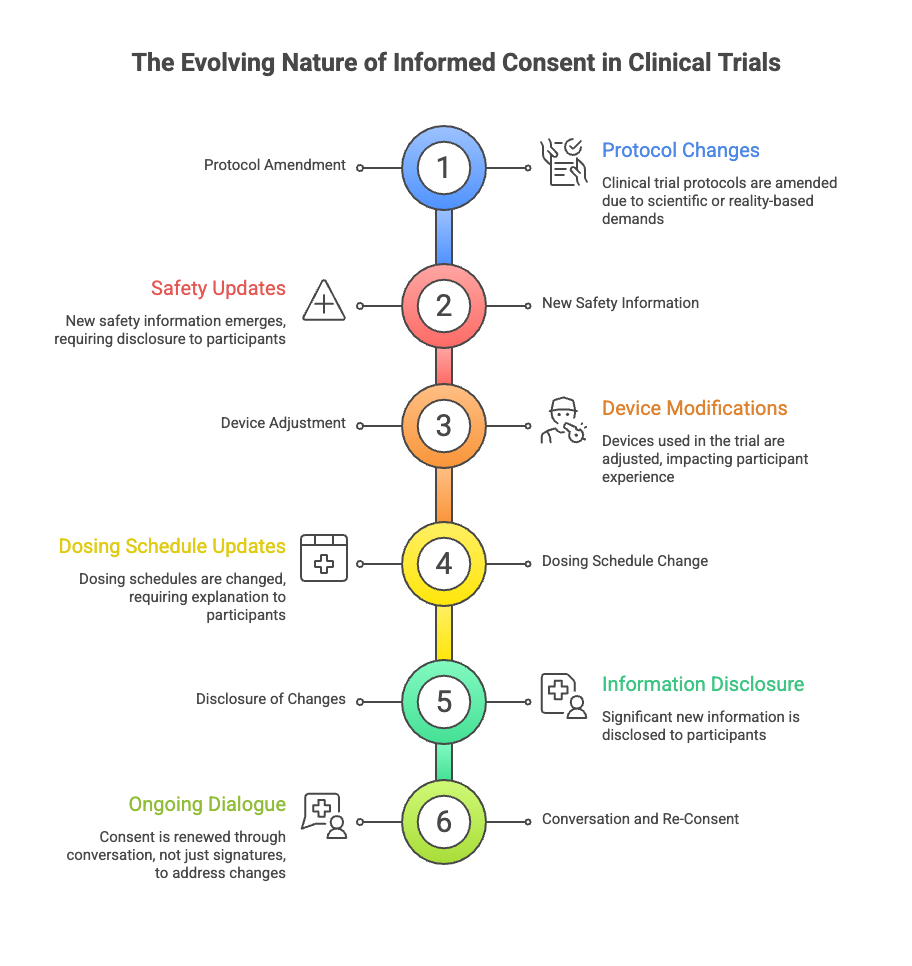
When the Story Changes Midway Through
Clinical trials are not static.
Protocols are amended. New safety information emerges. Devices are adjusted. Dosing schedules change. Sometimes the science demands it. Sometimes reality does.
Each change reshapes the experience for people already participating.
Regulations require that significant new information be shared. But disclosure alone doesn’t answer the question participants are really asking: What does this change mean for me now?
Good consent does not assume that a decision made once holds forever. It creates space to revisit expectations, to explain what has changed, and to acknowledge that continued participation is still a choice.
Consent, in practice, is renewed — not just with another signature, but with conversation.
Trust Isn’t Given Up Front
We often talk about informed consent as a threshold — a gate that must be passed before research can begin.
But trust is not built at thresholds. It’s built in the middle.
It grows when uncertainty is named rather than minimized. When questions are welcomed after the fact. When participants feel oriented, not just informed.
Seen this way, informed consent isn’t about securing agreement. It’s about staying accountable — long after the paperwork is done.
Understanding consent as a process doesn’t weaken trust in clinical research. It strengthens it.
Because trust, like consent, isn’t something people give once.
It’s something they continue to choose.

🧭 Compass Insight
Informed consent isn’t just about signing a form before a study begins.
It’s how people stay informed and supported while doctors and researchers are still learning what works.
When consent is treated as an ongoing conversation — not a one-time decision — people stay oriented, and trust has room to grow, even when the answers aren’t clear yet.
Informed consent is often treated as paperwork that allows research to move forward. In reality, it is the way people stay oriented while medicine is still learning.
Understanding that difference doesn’t just change how trials are run — it changes how we decide whether to take part.
References & Further Reading
Regulatory requirements for informed consent in clinical research are established by U.S. and international ethics and regulatory frameworks, including:
• U.S. FDA – 21 CFR Part 50: Protection of Human Subjects
• ICH E6(R2) Good Clinical Practice (GCP)
International guideline governing ethical and scientific standards for the design and conduct of clinical trials
Research shows that informed consent documents are often difficult for the general public to read and understand:
• FDA, “Informed Consent Information Sheet: Guidance for IRBs, Clinical Investigators, and Sponsors”
• NIH, “Clear Communication: An NIH Health Literacy Initiative”
These sources reflect ongoing efforts to improve how information is communicated to research participants while recognizing that documentation alone cannot replace meaningful, ongoing dialogue.